Retinitis pigmentosa (RP) is a rare, hereditary disease that causes the rod photoreceptors in the retina to gradually degenerate. The rods are located in the periphery of the retina and are responsible for peripheral and night vision. Cones, another type of photoreceptor, are densely concentrated in the macula. The cones are responsible for central visual acuity and color vision.
Rather than being considered a single disease, retinitis pigmentosa instead is viewed as a group of diseases affecting how light-sensitive cells in the back of the eye function. Not much is known about what causes retinitis pigmentosa, except that the disease is inherited.
The eye condition is associated with at least 32 different genes, which control traits that are passed along in a number of different ways. At times, the genetic trait is dominant and likely to be passed along to a child when a parent has RP. At other times, the trait for retinitis pigmentosa is recessive and may be present for many generations before it appears in a family member.
This means that, even if your mother and father don't have retinitis pigmentosa, you can still have the eye disease when at least one parent carries an altered gene associated with the trait. In fact, about 1 percent of the population can be considered carriers of genetic tendencies for retinitis pigmentosa.
Retinitis pigmentosa occurs in about 1 of every 4,000 people in the United States. When the trait is dominant, it is more likely to show up when people are in their 40s. When the trait is recessive, it tends to first appear when people are in their 20s.

The disease may be X-linked (passed from a mother to her son), autosomal recessive (genes required from both parents) or autosomal dominant (gene required from one parent) trait. Since it is often a sex-linked disease, retinitis pigmentosa affects males more than females.
People with RP usually first notice difficulty seeing in dim lighting and gradually lose peripheral vision. The course of RP varies. For some, the affect on vision may be mild. Others experience a progression of the disease that leads to blindness.
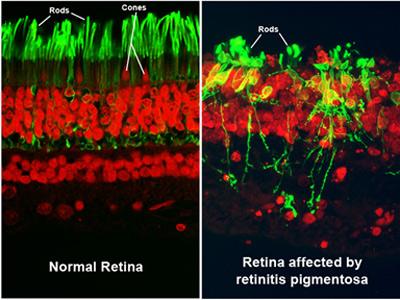
In many cases, RP is diagnosed during childhood when the symptoms begin to become apparent. However, depending on the progression of the disease, it may not be detected until later in life.


Rod and cone photoreceptor cell death in retinitis pigmentosa. Rod cell death due to the deleterious genetic mutations is associated with apoptosis, which involves the activation of caspase-independent pathways including poly-ADP-ribose-polymerase (PARP), calpain and histone deacetylase (HDAC). Cone cell death is induced by the microenviromental changes subsequent to rod degeneration, such as oxidation, inflammation and loss of trophic factors. Dying cones show different morphological features from rod cells, such as necrotic cytoplasmic swelling (asterisk), and is partly mediated through the activation of RIP kinase (RIPK). Electron microscopy images were reproduced with permissions from Murakami et al.5 mToR, mammalian target of rapamycin; RdCVF, rod-derived cone viability factor; TNF-α, tumor necrosis factor-α
SIGNS AND SYMPTOMS
Some of the most common symptoms of retinitis pigmentosa include:
- Decreased vision at night or in low light
- Loss of side (peripheral) vision, which may cause the person to bump into tables, furniture, or doorways. It may not be noticed by the person with retinitis pigmentosa, but may be apparent to others.
- Loss of central vision (in advanced cases)
- Other indicators of retinitis pigmentosa are your family history (especially the possibility of retinitis pigmentosa appearing in other family members) and expressed visual concerns or complaints, such as not being able to see well at night or in low light conditions.
- Difficulty seeing dim lighting
- Tendency to trip easily or bump into objects when in poor lighting
- Gradual loss of peripheral vision
- Glare
- Loss of contrast sensitivity
- Eye fatigue (from straining to see)
The initial retinal degenerative symptoms of retinitis pigmentosa are characterized by decreased night vision (nyctalopia) and the loss of the mid-peripheral visual field. The rod photoreceptor cells, which are responsible for low-light vision and are orientated in the retinal periphery, are the retinal processes affected first during non-syndromic forms of this disease.Visual decline progresses relatively quickly to the far peripheral field, eventually extending into the central visual field as tunnel vision increases. Visual acuity and color vision can become compromised due to accompanying abnormalities in the cone photoreceptor cells, which are responsible for color vision, visual acuity, and sight in the central visual field.The progression of disease symptoms occurs in a symmetrical manner, with both the left and right eyes experiencing symptoms at a similar rate.
A variety of indirect symptoms characterize retinitis pigmentosa along with the direct effects of the initial rod photoreceptor degeneration and later cone photoreceptor decline. Phenomena such as photophobia, which describes the event in which light is perceived as an intense glare, and photopsia, the presence of blinking, swirling or shimmering lights within the visual field, often manifest during the later stages of RP. Findings related to RP have often been characterized in the fundus of the eye as the "ophthalamic triad".
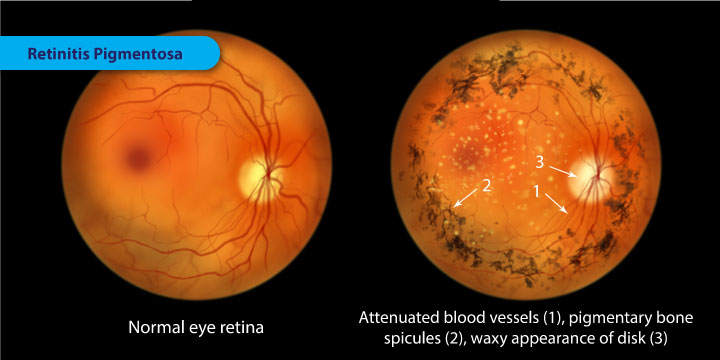
This includes the development of (1) a mottled appearance of the retinal pigment epithelium (RPE) caused by bony spicule formation, (2) a waxy appearance of the optic nerve, and (3) the attenuation of blood vessels in the retina.
Non-syndromic RP usually presents a variety of the following symptoms:
- Night blindness
- Tunnel vision (due to loss of peripheral vision)
- Latticework vision
- Photopsia (blinking/swirling/shimmering lights)
- Photophobia (aversion to bright lights)
- Development of bone spicules in the fundus
- Slow adjustment from dark to light environments and vice versa
- Blurring of vision
- Poor color separation
- Loss of central vision
- Eventual blindness
DETECTION AND DIAGNOSIS
It is a painless test. The ERG, in conjunction with the visual field exam, will usually make the diagnosis. The ERG will also determine if there is any involvement of the central retina and visual field. Periodic follow-up ERG examinations are necessary to follow and track the progression of your retinitis pigmentosa.
Retinitis pigmentosa is usually diagnosed before adulthood. It is often discovered when the patient complains of difficultly with night vision. The doctor diagnoses RP by examining the retina with an ophthalmoscope. The classic sign of RP is clumps of pigment in the peripheral retinal called "bone-spicules." A test called electroretinography (ERG) may also be ordered to study the eye's response to light stimuli. The test gives the doctor information about the function of the rods and cones in the retina. eResearch by Navid Ajamin -- spring 2012

Color fundus photograph of a patient with typical RP. There is peripheral bone spicule deposition encroaching into the macula, optic nerve pallor, and prominent vascular attenuation. The central retina and RPE are preserved (“central island”).

OCT B-scan corresponding to the Color fundus photograph . There is significant thinning of the outer retinal layers and dropout of the RPE involving the edges of the macula. However, the central fovea is spared with normal retinal architecture.
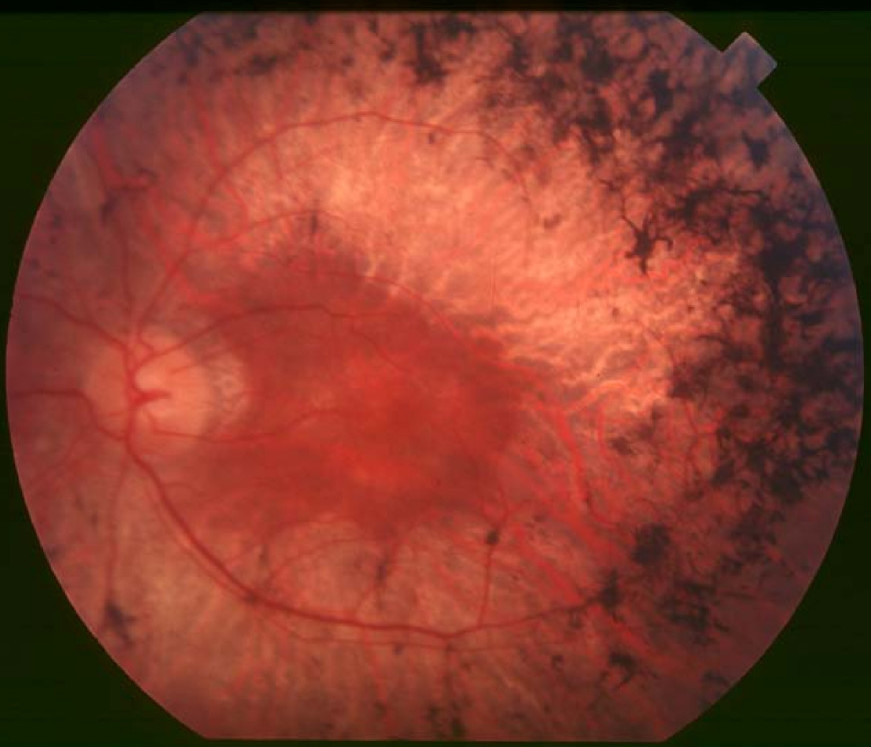
Retinitis pigmentosa (RP) refers to a heterogeneous group of inherited disorders that are characterized by loss of retinal cell function, preferentially in the peripheral retina. RP can have varying severity, age of onset, mode of inheritance, and systemic associations. RP may be inherited in an autosomal dominant, autosomal recessive, or X-linked recessive fashion. The X-linked form of the disease is typically the most severe. The disease is often secondary to mutations in the rhodopsin gene, though some forms have been linked to mutations in the RDS gene (Anasagasti et al., 2012). Generally, RP is characterized by a slowly progressive loss of night vision (nyctalopia) along with contraction of the visual field. In later stages of the disease central acuity is affected, which may cause profound vision loss. Typical fundus abnormalities include waxy pallor of the optic nerve, a tapetal-like reflex resulting from changes in the retinal pigment epithelium (RPE), narrowing of the peripheral retinal vasculature, and bone-spicule changes in the mid-peripheral retina. Definitive diagnosis requires electrophysiologic testing. Computed tomography is useful to aid in the initial diagnosis and detecting associated macular abnormalities such as cystoid macular edema.
Genetic Testing
Recently, testing for genetic defects is being done to clarify the loss in more detail and to find a treatment.
It is important to make a diagnosis so that the patient and family can be counseled as to the status of the disease, when driving might have to be discontinued, and what low vision interventions and low vision devices (in the case of more advanced disease) might be available to allow maximum use of the patient's visual potential.
TREATMENT
A group of eye problems that affect the retina, retinitis pigmentosa (RP) is a rare group of hereditary diseases that causes the photoreceptors in the retina to gradually degenerate.
People with retinitis pigmentosa lose their vision slowly over time, as it changes how the retina responds to light, making it hard to see.
The type and speed of vision loss from retinitis pigmentosa varies from person to person – it depends on their form of the condition.
There is currently no standard treatment or therapy for retinitis pigmentosa; however, scientists have isolated several genes responsible for the disease. Once RP is discovered, patients and their families are encouraged to seek genetic counseling.
There are few treatment options such as light avoidance and/or the use of low-vision aids to slow down the progression of RP. Some practitioners also consider vitamin A as a possible treatment option to slow down the progression of RP. Research suggests taking high doses of vitamin A (15,000 IU/day) may slow progression a little in some people, but the results are not strong. Taking too much vitamin A can be toxic and the effects of vitamin A on the disease is relatively weak. More research must be conducted before this is a widely accepted form of therapy.
Research is also being conducted in areas such as gene therapy research, transplant research, and retinal prosthesis. Since RP is usually the result of a defective gene, gene therapy has become a widely explored area for future research. The goal of such research would be to discover ways healthy genes can be inserted into the retina. Attempts at transplanting healthy retinal cells into sick retinas are being made experimentally and have not yet been considered as clinically safe and successful. Retinal prosthesis is also an important area of exploration because the prosthesis, a man-made device intended to replace a damaged body part, can be designed to take over the function of the lost photoreceptors by electrically stimulating the remaining healthy cells of the retina.Through electrical stimulation, the activated ganglion cells can provide a visual signal to the brain. The visual scene captured by a camera is transmitted via electromagnetic radiation to a small decoder chip located on the retinal surface. Data and power are then sent to a set of electrodes connected to the decoder. Electrical current passing from individual electrodes stimulate cells in the appropriate areas of the retina corresponding to the features in the visual scene.

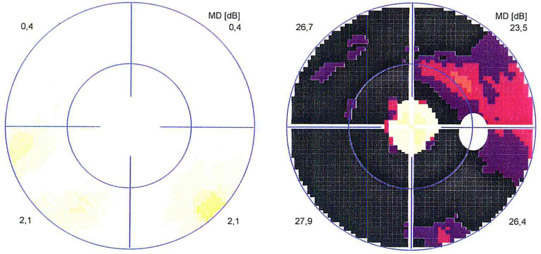
Visual field examination: Left: normal findings, right: tubular visual field loss in retinitis pigmentosa
Reference:
- augenklinik-sulzbach.de/patienten/51-behandlungsspektrum/netzhaut-chip/255-was-ist-retinitis-pigmentosa
- visionaware.org/info/your-eye-condition/retinitis-pigmentosa/how-is-retinitis-pigmentosa-diagnosed/125
- sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology/retinitis-pigmentosa
- eyedoctorpmb.co.za/eye-conditions/retinitis-pigmentosa
- allaboutvision.com/en-in/conditions/retina-pigmentosa
- stlukeseye.com/conditions/RetinitisPigmentosa.html
- genome.gov/Genetic-Disorders/Retinitis-Pigmentosa
- en.wikipedia.org/wiki/Retinitis_pigmentosa
See also:
- Groundbreaking Gene Therapy for Retinitis Pigmentosa
- Retinal Microchip Restores Vision in Retinitis Pigmentosa
- Detection of Retinitis Pigmentosa by Differential Interference Contrast Microscopy
 وبلاگ تخصصی عینک شامل مجموعه مطالب پزشکی است که اطلاعات مفیدی در رابطه با عینک , چشم، لنز، سلامتی چشم و راه های پیشگیری از بیماریهای چشمی، کنترل و درمان آن را در اختیار شما کاربر محترم می گزارد.
وبلاگ تخصصی عینک شامل مجموعه مطالب پزشکی است که اطلاعات مفیدی در رابطه با عینک , چشم، لنز، سلامتی چشم و راه های پیشگیری از بیماریهای چشمی، کنترل و درمان آن را در اختیار شما کاربر محترم می گزارد.